Déficit en acyl-coenzyme A déshydrogénase des acides gras à chaîne très longue
Déficit en acyl-coenzyme A déshydrogénase des acides gras à chaîne très longue
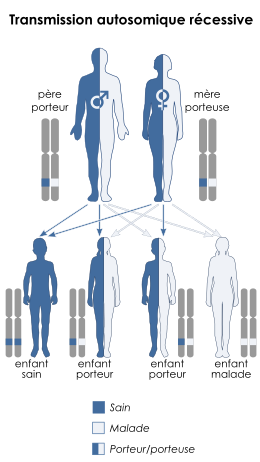
Le déficit en Acyl-Coenzyme A déshydrogénase des acides gras à chaîne très longue suit le principe de transmission par mode autosomique récessive. OMIM = 201475
| CIM-9 | 277.85 |
|---|---|
| OMIM | 201475 |
 Mise en garde médicale
Mise en garde médicale
modifier - modifier le code - voir Wikidata (aide) 
Le déficit en acyl-coenzyme A déshydrogénase des acides gras à chaîne très longue (en anglais Very long-chain acyl-coenzyme A dehydrogenase deficiency ou VLCADD) est un trouble de l'oxydation des acides gras qui empêche le corps de convertir certaines graisses en énergie, notamment pendant les périodes de jeûne[1].
Les personnes touchées par ce trouble ont un niveau insuffisant d'une enzyme permettant de décomposer un groupe de graisses appelées à très longue chaîne d'acides gras[2].
La génétique
Des mutations dans le gène ACADVL entraînent une insuffisance des niveaux d'enzyme appelée acyl-coenzyme A (CoA) déshydrogénase à chaîne très longue[3]. Sans cette enzyme, les chaînes d'acides gras très longues provenant de la nourriture et des graisses stockées dans le corps ne peuvent pas être dégradés et transformés. En conséquence, ces acides gras ne sont pas convertis en énergie, ce qui peut conduire à des signes et des symptômes de ce trouble, comme la léthargie et l'hypoglycémie. Des niveaux élevés d'acides gras à chaîne très longue ou partiellement dégradées en acides gras peuvent alors s'accumuler dans les tissus et peuvent endommager le cœur, le foie et les muscles, provoquant des complications plus graves[4].
Diagnostic
En général, les premiers signes et symptômes de ce trouble se produisent au cours de l'enfance et peuvent inclure :
- Faibles niveaux de sucre dans le sang (hypoglycémie)
- Le manque d'énergie (léthargie)
- Des faiblesses musculaires
- Des accès de rhabdomyolyse
Il y a également un risque élevé de complications telles que des anomalies dans le foie et des problèmes cardiaques sévères[4]. Les symptômes qui commencent plus tard dans l'enfance, l'adolescence ou l'âge adulte ont tendance à être plus doux et n'impliquent généralement pas de problèmes cardiaques. Les épisodes où ces sympthômes apparaissent sont généralement des périodes de jeûne, de la maladie ou d'exercice intense[5].
Certains autres premiers symptômes d'une crise métabolique sont : une forte somnolence, les changements de comportement, l'irritabilité de l'humeur, le manque d'appétit.
Certains de ces autres symptômes de VLCADD chez les nourrissons peuvent également comprendre de la fièvre, des nausées, de la diarrhée, des vomissements et l'hypoglycémie.
Traitement
Si une crise métabolique n'est pas traitée, un enfant avec la VLCADD peut développer des problèmes respiratoires, des convulsions, un coma, pouvant conduire à la mort[4].
La prise en charge de la déficience en VLCAD dépend de nombreux facteurs, y compris la forme de l'état et les signes et symptômes spécifiques présents. Par exemple, les personnes affectées par les formes sévères de l'état sont généralement obligées de suivre un régime faible en gras et à forte teneur en glucides avec des repas fréquents. Des calories supplémentaires peuvent être fournies par des triglycérides à chaîne moyenne (huile MCT). Si l'hospitalisation est nécessaire pour des épisodes aigus d'hypoglycémie et / ou de crise métabolique, du glucose intraveineux peut être administré en tant que source d'énergie. Les périodes de rhabdomyolyse peuvent être traitées avec une hydratation et une alcalinisation de l'urine (en diminuant la quantité d'acide prise) afin de protéger la fonction rénale et de prévenir une insuffisance rénale aiguë.
Les personnes affectées sont généralement conseillées d'éviter le jeûne, la déshydratation et un régime alimentaire riche en matières grasses[6].
Références
- ↑ Nancy D. Leslie, C. Alexander Valencia, Arnold W. Strauss et Jessica A. Connor, GeneReviews(®), Seattle (WA), University of Washington, Seattle, (PMID 20301763, lire en ligne)
- ↑ « NEWBORN SCREENING », sur www.newbornscreening.info (consulté le )
- ↑ (en) Genetics Home Reference, « VLCAD deficiency », sur Genetics Home Reference (consulté le )
- ↑ a b et c (en-US) « Very Long Chain Acyl CoA Dehydrogenase Deficiency (LCAD) - NORD (National Organization for Rare Disorders) », NORD (National Organization for Rare Disorders), 1996, 1998, 2001, 2004, 2010, 2013, 2016 (lire en ligne, consulté le )
- ↑ INSERM US14 -- TOUS DROITS RESERVES, « Orphanet: Déficit en acyl CoA déshydrogénase des acides gras à chaîne très longue », sur www.orpha.net (consulté le )
- ↑ (en) « VLCAD deficiency | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program », sur rarediseases.info.nih.gov (consulté le )
Liens externes
- La projection, de la Technologie Et de la Recherche en Génétique (STAR-G) Projet
v · m Maladie métabolique génétique du système lipidique : Troubles du métabolisme des acides gras (E71.3, 277.81–277.85) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Synthèse | |||||||||||||||||
| Dégradation |
| ||||||||||||||||
 Portail de la médecine
Portail de la médecine  Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire












